Author(s): Ruichu Gu, Xi Wang, Tianfan Fu.
Before you start
-
This tutorial assumes that you have already learned:
-
Basic biochemistry knowledge,
-
Basic Machine Learning knowledge;
-
-
Deeply explore this area you need to master the following:
-
Molecular Biology,
-
Computational structural biology,
-
Biochemistry,
-
Machine Learning & Deep Learning.
-
Introduction
What is RNA?
Ribonucleic acids (RNA) are polymeric molecules, assembled from a chain of nucleotides. Different from DNA, however, RNA is often single-stranded. An RNA molecule has a backbone made of alternating phosphate groups and the sugar ribose, rather than the deoxyribose found in DNA. Attached to each sugar is one of four bases: adenine (A), uracil (U), cytosine (C), or guanine (G).
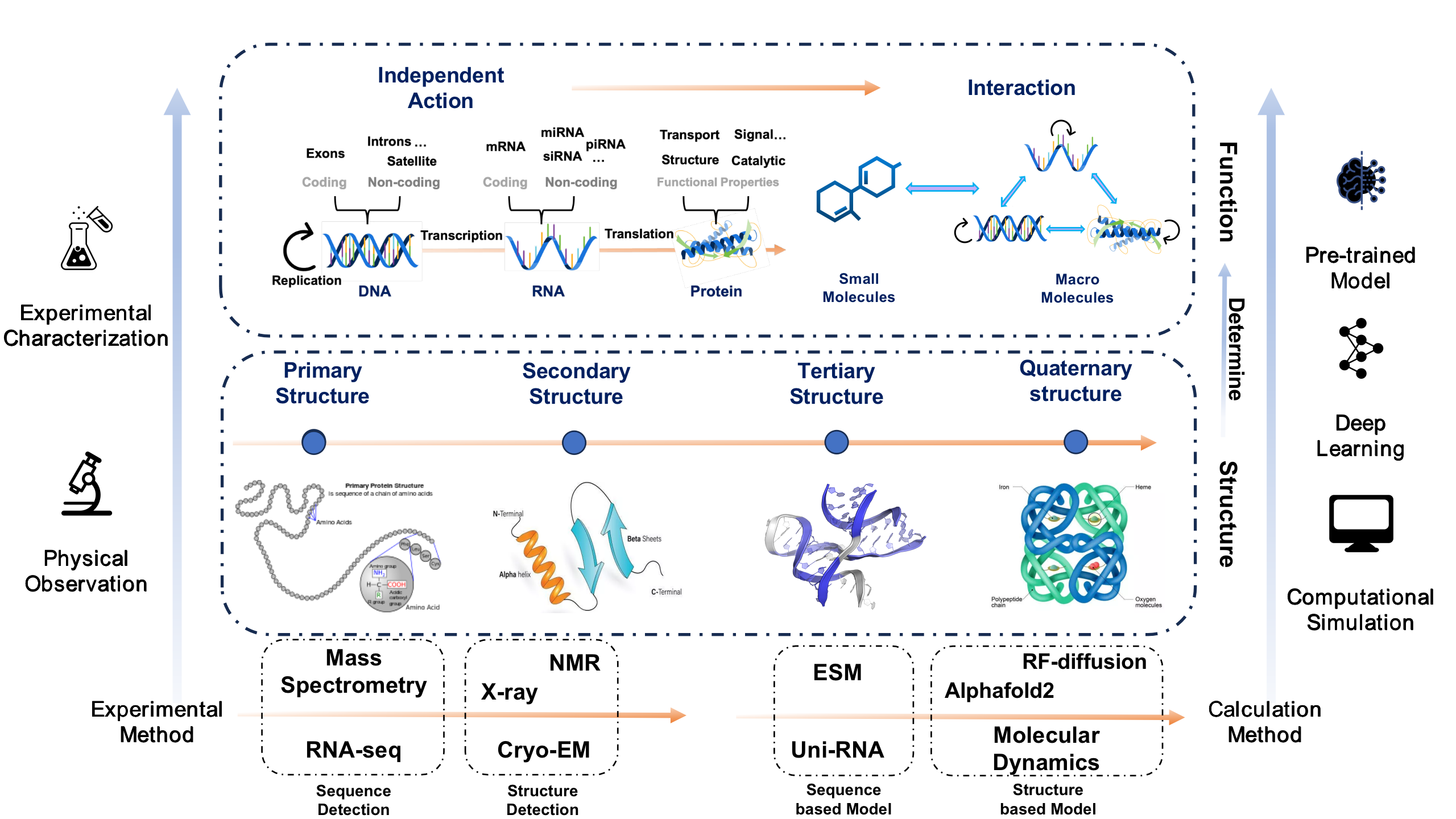
Here we give a simple framework to sort out the related work of computational structural biology in recent years. Based on the central dogma, we will introduce related studies on nucleic acids and proteins from the perspectives of structure and function, respectively. At the same time, we will also combine commonly used research methods in the field of structural biology, so that readers can have a general understanding of the research process of structural biology after reading. Readers can search for relevant information by themselves to further understand the subject background and its work.
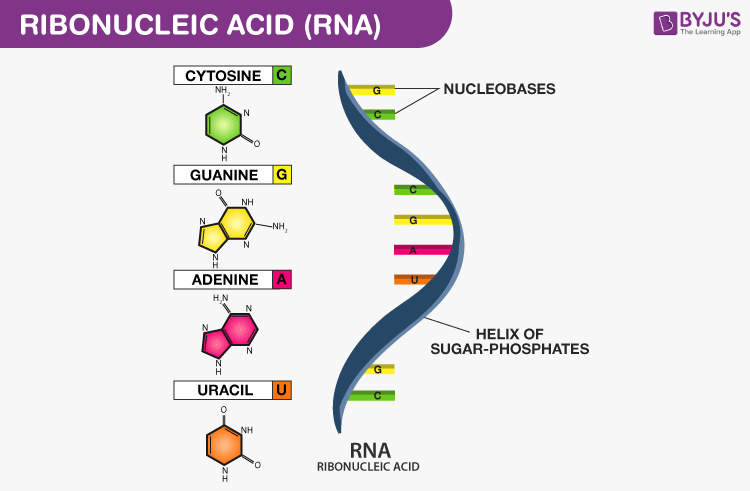
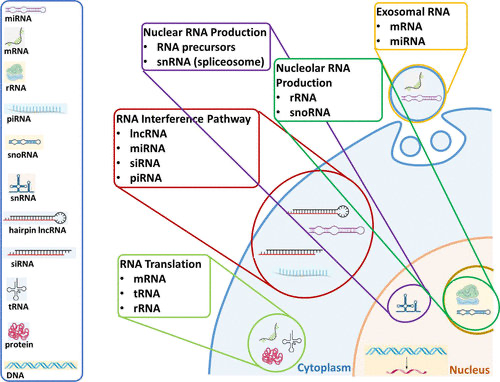
Each nucleotide in RNA contains a ribose sugar, with carbons numbered 1’ through 5’. A base is attached to the 1’ position, in general, adenine (A), cytosine (C), guanine (G), or uracil (U). A phosphate group is attached to the 3’ position of one ribose and the 5’ position of the next. The phosphate groups have a negative charge each, making RNA a charged molecule (polyanion). The bases form hydrogen bonds between cytosine and guanine, between adenine and uracil and between guanine and uracil. RNA plays an important role in biological processing (Figure 3). Different types of RNA exist in cells: messenger RNA (mRNA), ribosomal RNA (rRNA) and other non-coding RNA. Cellular organisms use messenger RNA (mRNA) to convey information from DNA to proteins, which is so-called coding RNA. Besides, non-coding RNA, containing no genetic information, participates in catalyzing biological reactions, controlling gene expression, or sensing and communicating responses to cellular signals through specialized structures.
Why Do We Study RNA?
Among the pathogenic proteins related to human diseases, more than 80%
of the proteins cannot be targeted by the current conventional small
molecule drugs and biological macromolecular preparations, which are
non-drugable protein targets. RNA-targeted drugs treat from the
transcriptional level, which can greatly expand the proportion of
therapeutic targets in the human genome. They have the advantages of
abundant candidate targets, short research and development cycle,
long-lasting drug efficacy, and high success rate of clinical
development. They are expected to become the third wave of modern new
drugs after drugs and antibody drugs.
RNA-targeted drugs are a completely new class of drugs that are
completely different from small-molecule drugs and antibody drugs. On
the one hand, they can target intracellular mRNA, ncRNA, etc., and
inhibit the expression of target proteins through gene silencing to
achieve the purpose of treating diseases; on the other hand, It is also
possible to develop a new generation of vaccines and protein replacement
therapy based on mRNA, which is expected to overcome diseases for which
there is no drug, including genetic diseases and other intractable
diseases. The future potential application scenarios of RNA-targeted
drugs are broad.
After nearly 40 years of development, oligonucleotide-based RNA-targeted
drug development has made significant progress in delivery technology,
stable chemical modification, pharmaceutical research, and clinical
translation, making the clinical effect of RNA-targeted drugs gradually
get verified. In August 2020, the world’s first small molecule RNA
targeting drug Evrysdi was approved by the FDA.
Why Do We Study RNA with AI/ML?
There are several reasons why AI and machine learning can be valuable tools in studying RNA:
-
RNA is a complex molecule: RNA molecules can have a variety of structures and functions, making it difficult to study them using traditional methods. AI and deep learning can be used to analyze large amounts of data and identify patterns that might not be noticeable through manual inspection.
-
Large amount of data: RNA-seq and other high-throughput sequencing technologies have generated a huge amount of data, making it difficult to analyze manually. AI and deep learning algorithms can help process and make sense of this data, reducing the time and effort required.
-
Predictive modeling: AI and deep learning can be used to make predictions about RNA structure and function based on sequence data. This can help researchers identify potential targets for drug development or understand the mechanisms of RNA-related diseases.
Current Status of RNA Research
RNA Structure Prediction
The structural features of RNA are closely related to its physiological functions. RNA secondary structure and tertiary structure prediction are popular research directions at present.
RNA Secondary Structure
Secondary structure plays an important role in determining the function of noncoding RNAs. Hence, identifying RNA secondary structures is of great value to research. Over the years, scientists have developed a set of tools for secondary structure prediction, where remarkable progress has been achieved. However, the problem is still unsolved until now. This section will give a brief introduction to RNA secondary structure and related machine learning algorithms.
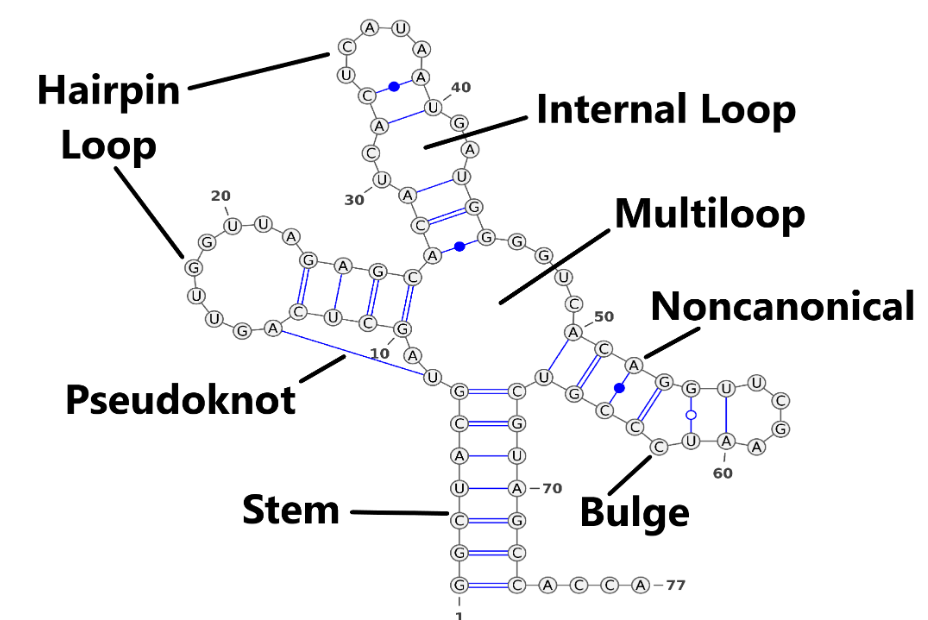
Similar to proteins, RNA forms secondary structures through hydrogen bonds between bases. Stem is the most important component of RNA secondary structure, and all other secondary structures, except pseudo-knots, can be deduced from stems. For example, Internal Loop is a combination of two stems. See Figure 4. For representation of the secondary structure, we have three widely used methods.
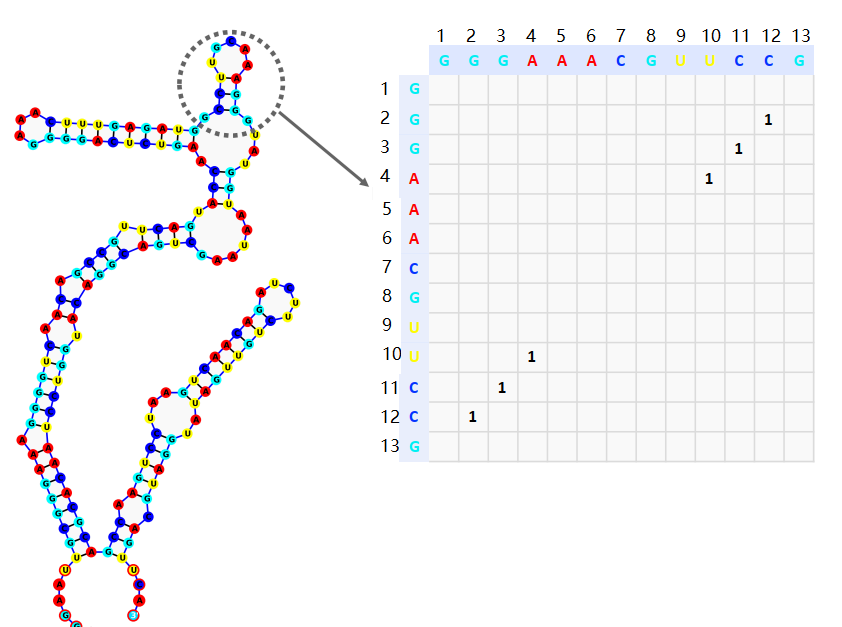
Adjacency matrix
. The adjacency matrix consisting of 0 and 1. \(a_{ij=1}\) denotes the base pairing at the \(i\)th and \(j\)th positions on the sequence to form a hydrogen bond (Figure 5). Here the adjacency matrix is a perfectly symmetric matrix.
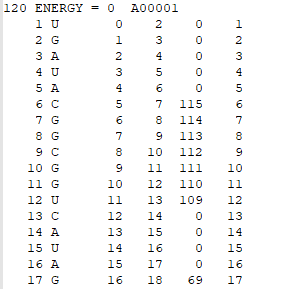
Connectivity Table
The first line of the CT file includes the total number of nucleotides, the folding energy, and the file name. In the other rows, each row contains 6 columns of data, which are: i) List of the nucleotides from 1 to N (N = total number of nucleotides); ii) List of the type of nucleotide (A, G, U, or C); iii) List of the nucleotides increasing from zero to N - 1; iv) List of the nucleotides from 2 to N; v) List of the nucleotides that are paired to those listed in increasing order. Any zeros in the fifth column indicate that the particular nucleotide is unpaired; vi) A repeat of column i.
Dot Bracket Annotation
The unpaired bases in the RNA sequence are denoted by ".", and the two bases that form hydrogen pairs are denoted by "(" and ")". Bases in the base pair relatively close to the 5’ end are denoted by "(", and bases in the base pair relatively close to the 3’ end are denoted by ")".

For researchers, RNA secondary structure prediction can be viewed as multi-classification or pixel prediction problems. In recent years, there have been lots of excellent work that focus on these challenges. Even though more mature RNA secondary structure datasets are now available, their total sample size is still relatively small. Commonly used datasets and their URLs are listed in Table 1.
| Database | URLs |
|---|---|
| bpRNA | bpRNA Website |
| RNAStralign [2] | RNAStralign GitHub |
| ArchiveII [3] | ArchiveII GitHub |
We already have lots of strategies for RNA representation. The simplest one is using one hot encoding. For ‘A’, ‘U’, ‘C’, and ‘G’ bases, four dimensions coding is enough. Besides, under IUPAC standard, more codes are used (Table 2).
| IUPAC nucleotide code | Base |
|---|---|
| A | Adenine |
| C | Cytosine |
| G | Guanine |
| T (or U) | Thymine (or Uracil) |
| R | A or G |
| Y | C or T |
| S | G or C |
| W | A or T |
| K | G or T |
| M | A or C |
| B | C or G or T |
| D | A or G or T |
| H | A or C or T |
| V | A or C or G |
| N | any base |
| . or - | gap |
Besides one hot encoding, there are other methods of representations. Multiple Sequence Alignment (MSA) is a vital method for genetic study, which contain co-evolutionary information. The AlphaFold2 and other models have proved the power of MSA for protein structure prediction. For RNA, we can also use MSA as model inputs. 2D features generated from MSA are also useful, for examples, Position-specific scoring matrix (PSSM), GREMLIN Matrix, and plmDCA. Some probability matrix calculated by free-energy based algorithms can also be used as input features, for instance, LinearFoldPartition provides rapid and accurate calculation of bases-pair probability matrix. Very recently, Li. et al. reported a large pre-trained models, which gives sequence embedding with 640 dimensions [4]. Latent embedding from pre-trained models are also powerful. Commonly-used features for RNA are listed in Table 3.
| Features | Descriptions |
|---|---|
| One-hot Encoding | Discrete feature encoding in Euclidean space |
| Probability matrix | Base pairing probability matrix calculated based on thermodynamic principle |
| MSA | Multi-species sequence similarity relationships |
| PSSM | Sequence position-specific base frequencies |
| GREMLIN | Markov random field-based graph model |
| Pre-trained Embedding | Latent sequences embedding |
1D Approaches
The 1D approaches treat RNA secondary structure as sequences. For instance, by using dot-bracket annotation as target, researchers convert problems into multi-classification tasks. The ".", "(", and ")" can be viewed as three classes. This is a traditional "seq2seq" problems. Researchers can use relevant models from the field of Natural Language Processing (NLP) to solve this problem, such as RNN, LSTM, and Transformer. 1D approaches use a loss function similar to that of classification problems, such as cross-entropy. Class imbalance may occur for such classification problems due to the fact that the number of paired bases on an RNA sequence is often less than the number of unpaired bases.
2D Approaches
The 2D approach focuses on the adjacency matrix as the prediction target, which is an \(L*L\) two-dimensional matrix, where each element value represents the base pairing probability. The 2D approach for RNA secondary structure prediction has a strong similarity to the semantic segmentation task in computer vision. Researchers can use similar ideas to study RNA secondary structure prediction problems. For example, Ufold [5] used the classical Unet model in semantic segmentation and achieved excellent results even with simple features.
RNA Tertiary Structure
The tertiary structure prediction of RNA is similar to that of proteins, again trying to obtain the 3D coordinates of the atoms. However, the tertiary structure prediction of RNA is still dominated by molecular simulation methods, and machine learning methods are still in a relatively rudimentary stage, mainly due to the lack of a large amount of high-precision tertiary structure data at present. Even though molecular simulation methods are well developed, they still consume long computational time and are moderately accurate. It is worth noting that molecular simulation methods are able to obtain tertiary structures with high accuracy if high-precision secondary structures are used as constraints. In addition, borrowing ideas from earlier protein structure prediction, using machine learning methods to obtain contact maps as fuzzy inputs to molecular simulation algorithms is also a feasible approach. In addition, Li and colleagues proposed an end-to-end structure prediction model E2Efold-3D. Table 4 lists some machine learning methods for RNA tertiary structure prediction.
| Models | Authors | Date |
|---|---|---|
| Euclidean distance neural networks [1] | Nikolay V. Dokholyan | 2022 |
| Geometric Potentials from Deep Learning [2] | Yang Zhang | 2022 |
| E2Efold-3D: End-to-End Deep Learning Method [3] | Yu Li | 2022 |
Interaction of RNA with other substances
RNA Binding Protein
RNA Binding proteins are key regulators of cellular and developmental
processes and play a key role in post-transcriptional splicing and
localization of mRNA. Defining the properties of RBP will allow us to
regulate the protein better. RBP is usually combined with specific
motifs in RNA structures through RNA Binding domains.
At present, the most definitive way to study RNA binding to proteins is
to extract them from high-resolution experimental structures of
protein-RNA complexes, which is expensive and time-consuming. Therefore,
it is of great significance to develop available computational methods
to identify Binding information in RNA Binding proteins.
RNA with Small Molecule
The increasing appreciation for the crucial roles of RNAs in infectious
and non-infectious human diseases makes them attractive therapeutic
targets. Coding and non-coding RNAs frequently fold into complex
conformations which, if effectively targeted, offer opportunities to
therapeutically modulate numerous cellular processes, including those
linked to undruggable protein targets. RNA is a legitimate target of
small molecules and that ligands can indeed bind discrete binding
pockets in RNA, cementing the applicability of small-molecule treatment
to target RNAs.
Overall, three major components are required to support the development
of efficacious RNA-based small molecule therapeutics: (1) the
identification of therapeutically valuable RNA target, (2) the
development of a screening method that will establish drug-like
molecules against target RNA, and (3) the identification of RNA motifs
that accommodate ligand binding with high affinity and specificity.
Current ML techniques can be used to screen small molecules and evaluate
the binding specificity of small molecules, which can improve the
selection of small molecule structures with RNA affinity from a large
number of unknown targets.
RNA Sequence Optimization
The epidemic of COVID-19 has promoted the research and development of RNA vaccines. In addition to the encapsulation and delivery system, the selection of appropriate RNA sequences can enable the vaccine to be expressed in host cells more efficiently. At present, the relevant optimization studies mainly focus on prokaryotes. Due to the complex transcription-translation mechanism of eukaryotes, there is no good optimization method.
RNA Function Prediction
RNA function prediction refers to the task of exploring the biological function of RNA. In particular, non-coding RNAs, which play regulatory and catalytic roles in cells by binding to other macromolecules such as proteins, as well as RNAs that are beacons for certain diseases, are important research topics in medical research. In this article, we list some common RNA function prediction tasks for which machine learning techniques may bring revolutionary changes.
Protein-RNA interaction
RNA-protein interactions is crucial to discovery the mechanisms and control of gene expression and biological synthesis. Traditional molecular simulation methods may suffer from low accuracy and long computing time. With the development of machine learning technology in recent years, this classical field may welcome revolution.
mRNA untranslated region’s function
Untranslated Region (UTR) refers to the region on the mRNA that does not carry genetic information, which is usually at both ends of the Coding Sequence (CDS) region. Biologists have found that specific sites on it play an important role in gene expression and regulation. Therefore, the delicate identification of the UTR region can help biologists to discover new regulatory sites for pathogenesis and drug development.
Analysis of single-cell RNA sequencing data
Single-cell RNA sequencing (scRNA-seq) refers to profile the whole transcriptome of a large number of individual cells. scRNA-seq helps biologists to uncover gene expression information of single cells more precisely, i.e. to identify expression differences between different cells in the same tissue. However, due to the huge amount of data it generates, data processing is a more difficult step in scRNA-seq. Machine learning methods are good at extracting knowledge from huge amount of information, so applying ML in this field is a promising approach.
Interaction between LncRNA and RNA binding protein
Up to now, the majority of Long non-coding RNA (LncRNA) functions are mediated by lncRNA-protein interactions. lncRNAs perform diverse functions by forming RNA-protein complexes with proteins, such as chromosomal regulatory complexes, transcription factors and ribonucleoprotein (RNP) complexes. Therefore, techniques to study LncRNA-protein interactions can help us unravel the biological functions of LncRNAs.
Outlook: AI and RNA Study
Problems are getting harder for scientists to solve due to the enormity of the data that needs processing in the big data era. It’s tough for researchers to catch the ‘Big Pictures’. The treasure of RNA and RNA-related drugs are hidden in the intricate books of genes and cells. Artificial Intelligence (AI) technique is expert at exploring that high dimensional puzzles. It looks promising that applying AI in RNA sciences. Here we will give two successful examples for AI and RNA.
Successful Cases
- ARES
In 2021, Stanford University researcher reported a new deep-learning system called Atomic Rotationally Equivariant Scorer (ARES) significantly improves the prediction of RNA structures over previous artificial intelligence (AI) models. Although RNA molecules fold into intricate 3D structures like proteins that allow them to perform a wide range of cellular functions, there is far less training data available for RNA structures prediction. To address this problem, ARES accept simple 3D coordinates and chemical element type of each atom as input with less preprogrammed assumptions. With FARFAR2, ARES achieved excellent performance on RNA puzzles dataset. The clever design of the neural network guaranteed generalizing ability. Besides, start from physical view is also critical. Learning from the essential properties of the sturctures makes it convincing and reasonable.
- RNA-Binding Protein
Interactions with RNA-binding proteins (RBPs) are integral to RNA function and cellular regulation, and dynamically reflect specific cellular conditions. However, current methods can’t predict dynamic RBP binding in various cellular conditions. By using in vivo data and deep learning models, Researchers from Tsinghua University developed a power AI tools for accurate prediction of RBP binding problems. The rich profiling data and deep learning-based prediction tool provide access to a previously inaccessible layer of cell-type-specific RBP–RNA interactions, with clear utility for understanding and treating human diseases. One of the most obstacles of RBP would be how to extract rules and knowledge from millions to billions of samples. Fortunately, handing large-scale data is the speciality of AI/ML. It’s promising the AI/ML will bring breakthrough progress.
Recommended Materials for Studying RNA with AI/ML
| Titles | Authors |
|---|---|
| Artificial Intelligence and Molecular Biology | Lawrence E. Hunter |
| Molecular Biology of RNA | David Elliott, Michael Ladomery |
| Deep Learning | Ian Goodfellow, Yoshua Bengio, Aaron Courville |
Protein Structure Prediction
The discovery of targets is greatly impacted by the three-dimensional structures of proteins, given that a protein’s biological role predominantly arises from its spatial configuration. Nobel laureate Dr. Christian Anfinsen proposed in 1972 that the 3D structure of a protein should be definitively determined by its sequence of amino acids [9]. In most instances, researchers initiate their work with knowledge of a protein’s amino acid sequence, which is derived from genetic information. Subsequently, they strive to ascertain its three-dimensional structure to gain deeper insights into its physical, chemical, and biological attributes. Conventional experimental techniques, such as X-ray crystallography [10], can precisely elucidate protein structures, yet these methodologies are resource-intensive and time-demanding. Out of the approximately 1 billion documented protein amino acid sequences, only around 100 thousand structures have been identified using these approaches, encompassing approximately 17% of human proteins [11].
Protein Structure Prediction, also known as protein folding, aims at predicting the 3D structure of protein given the amino acid sequence. This hypothesis ignited a fifty-year-long endeavor to predict protein 3D structures computationally using their amino acid sequence as a foundation.
What is Protein and Protein Structure?
Proteins are the building blocks of life. Every cell in the human body contains proteins. Proteins carry out all the essential functions in the human body. Proteins support almost all biological activities. For example, stomach enzymes serve to digest food; the movement of myosin on actin is the driving force for muscular contraction; whey proteins strengthen cell anti-oxidation; and antibodies protect us from disease.
The basic protein structure is a chain of amino acids. The amino acid chain can fold into a 3D structure.
Four levels of hierarchical structures usually represent a protein: (1) primary structure (i.e., amino acid sequence), (2) secondary structure, (3) tertiary structure, and (4) quaternary structure. They characterize the hierarchical structure of a protein at different levels of complexity, as described below.
-
Primary structure. The primary structure of a protein is simply the sequence of amino acids in a polypeptide chain. There are a total of 20 natural amino acids, as shown in Table 6.
-
Secondary structure. The secondary structure of a protein refers to regular, recurring arrangements in the space of adjacent amino acid residues in a polypeptide chain. It characterizes the localized conformation of the polypeptide chain. There are a total of 8 categories of secondary structures, e.g., \(\alpha\)-helix, Turn, Bend, \(\pi\)-helix, 3-10 helix, Strand, Isolated beta-bridge residue, and None (also called a coil, which is not in any above formations). Each amino acid belongs to one of these eight secondary structures.
-
Tertiary structure. The tertiary structure of a protein refers to the overall three-dimensional arrangement of its polypeptide chain in Euclidean space. An amino acid consists of an \(\alpha\) (central) carbon atom linked to an amino group, a carboxyl group, a hydrogen atom, and a variable component called a side chain. 3D graph structure is also known as the backbone structure, which belongs to the tertiary structure of a protein.
-
Quaternary structure. The quaternary structure of a protein describes the relationship of multiple polypeptide chains (or subunits) into a closely packed arrangement. Each polypeptide chain has its own primary, secondary, and tertiary structure. Hydrogen bonds and van der Waals forces between nonpolar side chains arrange the subunits together. It lies in the fourth and highest level of protein structure. The proteins with a single polypeptide chain do not have quaternary structures.
Protein Structure Prediction
We discuss some essential components for protein structure prediction.
-
Feature engineering.
-
Multiple sequence alignment (MSA) is a feature engineering technique to align three or more biological sequences (generally protein or DNA) and compare them for similarity. It is an essential tool in bioinformatics and is used in various applications, including phylogenetic tree construction, protein domain identification, protein structure and function prediction, and understanding evolutionary processes. MSA is used for most protein folding methods, including RaptorX-Contact [12], AlphaFold1 [13], trRoseTTA [14], AlphaFold2 [15], RoseTTAFold [16], AlphaFold-Multimer [17], UniFold [18], and OpenFold [19].
-
Language model (LM). The process of Multiple Sequence Alignment (MSA) is often characterized by its time-intensive and resource-heavy nature, primarily due to its computationally intensive approach. To address this challenge, ESMFold [20] offers a solution by employing a large-scale language model that is pre-trained on amino acid sequences. This approach replaces the conventional MSA with a robust neural representation, substantially accelerating the process.
-
-
Learning strategy. These are two kinds of learning strategies for the existing protein folding methods.
-
Two-stage learning. The earlier protein folding approaches leverage two-stage learning: (1) predicts pairwise distance and torsion angles and (2) constructs potential function based on the predicted distance and torsion angle and optimizes it with gradient descent. The strategy is used in RaptorX-Contact [12], AlphaFold1 [13], trRoseTTA [14].
-
End-to-end learning. End-to-end learning is a powerful learning approach that utilizes gradient-based optimization on the entire system, which can comprise various differentiable components such as neural network modules. This training strategy allows the system to learn rich representations directly from raw inputs, eliminating the need for manual feature engineering. The strategy is used in AlphaFold2 [15], RoseTTAFold [16], AlphaFold-Multimer [17], UniFold [18], and OpenFold [19].
-
-
Neural Network.
-
Residual Convolutional Neural Network. Residual CNN takes the MSA feature as input. The groundtruth of residual CNN is the distance matrix and torsion angle, which are translation- and rotation-invariant. Thus, it is \(SE(3)\)-invariant. It uses dilated Convolutional Neural Network, a variant of convolutional neural networks that incorporates dilated convolutions. Dilated convolutions introduce a dilation rate parameter that controls the spacing between sampled elements within the convolutional kernel, affecting the receptive field size of the output; e.g., a dilation rate of 2 means sampling every two elements. The main feature of dilated convolutions is the ability to increase the network’s receptive field, meaning each output unit can "see" a wider region of the input. Residual connection is used to alleviate gradient vanishment issues. Residual CNN is used in RaptorX-Contact [12], AlphaFold1 [13], trRoseTTA [14].
-
Evoformer. The Evoformer is a variant of the transformer architecture specifically designed to leverage evolutionary sequence information. It stacks 48 basic transformer blocks repeatedly with residual connection to alleviate gradient vanishment issues. There is no parameter sharing between these blocks. For each block, the input and output are of the same shapes. It is \(SE(3)\)-equivariant and used in AlphaFold2 [15], AlphaFold-Multimer [17], UniFold [18], ESMFold [20], and OpenFold [19].
-
Table 7 summarizes the different strategies for different SOTA protein folding methods. More information can be found at [21].
| Class | Abbreviation | Percentage (%) |
|---|---|---|
| Glycine | Gly / G | 7.6% |
| Alanine | Ala / A | 7.7% |
| Valine | Val / V | 7.0% |
| Leucine | Leu / L | 8.6% |
| Isoleucine | Ile / I | 5.5% |
| Proline | Pro / P | 4.6% |
| Phenylalanine | Phe / F | 3.6% |
| Tyrosine | Tyr / Y | 3.1% |
| Tryptophan | Trp / W | 1.2% |
| Serine | Ser / S | 6.7% |
| Threonine | Thr / T | 5.7% |
| Cystine | Cys / C | 1.3% |
| Methionine | Met / M | 2.7% |
| Asparagine | Asn / N | 4.1% |
| Glutamine | Gln / Q | 3.9% |
| Aspartic acid | Asp / D | 5.3% |
| Glutamic acid | Glu / E | 6.5% |
| Lysine | Lys / K | 6.3% |
| Arginine | Arg / R | 5.3% |
| Histidine | His / H | 3.1% |
This table summarizes existing protein folding approaches, including RaptorX-Contact [12], AlphaFold1 [13], trRoseTTA [14], AlphaFold2 [15], RoseTTAFold [16], AlphaFold-Multimer [17], UniFold [18], ESMFold [20], and OpenFold [19]. Two-stage learning typically consists of (1) prediction of pairwise distance and orientation and (2) energy minimization. The table is copied from Artificial Intelligence in Protein Folding [21].
| Methods | Feature | Learning | Network | Symmetry |
|---|---|---|---|---|
| RaptorX-Contact | MSA | Two-Stage | Residual CNN | (SE(3))-Invariant |
| AlphaFold1 | MSA | Two-Stage | Residual CNN | (SE(3))-Invariant |
| trRoseTTA | MSA | Two-Stage | Residual CNN | (SE(3))-Invariant |
| AlphaFold2 | MSA | End-To-End | Evoformer + Structure | (SE(3))-Equivariant |
| RoseTTAFold | MSA | End-To-End | Three-Track NN | (SE(3))-Equivariant |
| UniFold | MSA | End-To-End | Evoformer + Structure | (SE(3))-Equivariant |
| ESMFold | LM | End-To-End | Evoformer + Structure | (SE(3))-Equivariant |
| OpenFold | MSA | End-To-End | Evoformer + Structure | (SE(3))-Equivariant |
| AlphaFold-Multimer | MSA | End-To-End | Evoformer + Structure | (SE(3))-Equivariant |
References
[1] Xinshi Chen, Yu Li, Ramzan Umarov, Xin Gao, and Le Song. “RNA secondary structure prediction by learning unrolled algorithms.” arXiv preprint arXiv:2002.05810, 2020.
[2] Zhen Tan, Yinghan Fu, Gaurav Sharma, and David H Mathews. “Turbofold ii: RNA structural alignment and secondary structure prediction informed by multiple homologs.” Nucleic acids research, 45(20):11570–11581, 2017.
[3] Michael F Sloma and David H Mathews. “Exact calculation of loop formation probability identifies folding motifs in RNA secondary structures.” RNA, 22(12):1808–1818, 2016.
[4] Jiayang Chen, Zhihang Hu, Siqi Sun, Qingxiong Tan, Yixuan Wang, Qinze Yu, Licheng Zong, Liang Hong, Jin Xiao, Tao Shen, et al. “Interpretable RNA foundation model from unannotated data for highly accurate RNA structure and function predictions.” bioRxiv, 2022.
[5] Laiyi Fu, Yingxin Cao, Jie Wu, Qinke Peng, Qing Nie, and Xiaohui Xie. “Ufold: fast and accurate RNA secondary structure prediction with deep learning.” Nucleic acids research, 50(3):e14–e14, 2022.
[6] Congzhou Mike Sha, Jian Wang, and Nikolay V Dokholyan. “Predicting 3D RNA structure from solely the nucleotide sequence using euclidean distance neural networks.” bioRxiv, 2022.
[7] Robin Pearce, Gilbert S Omenn, and Yang Zhang. “De novo RNA tertiary structure prediction at atomic resolution using geometric potentials from deep learning.” bioRxiv, 2022.
[8] Tao Shen, Zhihang Hu, Zhangzhi Peng, Jiayang Chen, Peng Xiong, Liang Hong, Liangzhen Zheng, Yixuan Wang, Irwin King, Sheng Wang, et al. “E2efold-3d: End-to-end deep learning method for accurate de novo RNA 3D structure prediction.” arXiv preprint arXiv:2207.01586, 2022.
[9] John C Wooley and Yuzhen Ye. “A historical perspective and overview of protein structure prediction.” Computational Methods for Protein Structure Prediction and Modeling: Volume 1: Basic Characterization, pages 1–43, 2007.
[10] MS Smyth and JHJ Martin. “X ray crystallography.” Molecular Pathology, 53(1):8, 2000.
[11] Kathryn Tunyasuvunakool, Jonas Adler, Zachary Wu, Tim Green, Michal Zielinski, Augustin Žídek, Alex Bridgland, Andrew Cowie, Clemens Meyer, Agata Laydon, et al. “Highly accurate protein structure prediction for the human proteome.” Nature, 596(7873):590–596, 2021.
[12] Sheng Wang, Siqi Sun, Zhen Li, Renyu Zhang, and Jinbo Xu. “Accurate de novo prediction of protein contact map by ultra-deep learning model.” PLOS Computational Biology, 13(1):e1005324, 2017.
[13] Andrew W. Senior, Richard Evans, John Jumper, James Kirkpatrick, Laurent Sifre, et al. “Improved protein structure prediction using potentials from deep learning.” Nature, 577:706–710, 2020.
[14] Zongyang Du, Hong Su, Wenkai Wang, Lisha Ye, Hong Wei, Zhenling Peng, Ivan Anishchenko, David Baker, and Jianyi Yang. “The trrosetta server for fast and accurate protein structure prediction.” Nature Protocols, 16(12):5634–5651, 2021.
[15] John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. “Highly accurate protein structure prediction with AlphaFold.” Nature, 596(7873):583–589, 2021.
[16] Minkyung Baek, Frank DiMaio, Ivan Anishchenko, Justas Dauparas, Sergey Ovchinnikov, Gyu Rie Lee, Jue Wang, Qian Cong, Lisa N Kinch, R Dustin Schaeffer, et al. “Accurate prediction of protein structures and interactions using a three-track neural network.” Science, 373(6557):871–876, 2021.
[17] Richard Evans, Michael O’Neill, Alexander Pritzel, Natasha Antropova, Andrew Senior, Tim Green, Augustin Žídek, Russ Bates, Sam Blackwell, Jason Yim, et al. “Protein complex prediction with alphafold-multimer.” bioRxiv, pages 2021–10, 2021.
[18] Ziyao Li, Xuyang Liu, Weijie Chen, Fan Shen, Hangrui Bi, Guolin Ke, and Linfeng Zhang. “Uni-fold: an open-source platform for developing protein folding models beyond alphafold.” bioRxiv, pages 2022–08, 2022.
[19] Gustaf Ahdritz, Nazim Bouatta, Sachin Kadyan, Qinghui Xia, William Gerecke, Timothy J O’Donnell, Daniel Berenberg, Ian Fisk, Niccolò Zanichelli, Bo Zhang, et al. “OpenFold: Retraining alphafold2 yields new insights into its learning mechanisms and capacity for generalization.” bioRxiv, pages 2022–11, 2022.
[20] Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, et al. “Evolutionary-scale prediction of atomic-level protein structure with a language model.” Science, 379(6637):1123–1130, 2023.
[21] Xuan Zhang, Limei Wang, Jacob Helwig, Youzhi Luo, Cong Fu, Yaochen Xie, Meng Liu, Yuchao Lin, Zhao Xu, Keqiang Yan, et al. “Artificial intelligence for science in quantum, atomistic, and continuum systems.” arXiv preprint arXiv:2307.08423, 2023.