Physics
Molecular Dynamics
Molecular Dynamic (MD) is a type of molecular simulation method, which aims to study the dynamic evolution of physical systems through computer simulations of atoms and molecules. Based on MD simulations and statistical mechanics, many macroscopic thermodynamic properties, for instance, free energy or density, can be evaluated. Typically, trajectories of atoms in a simulation are generated by solving Newton’s laws of motion, where the potential energy function \(V\) comes either from force fields or Quantum Mechanic (QM) ab-initio calculations:
Depending on the smallest indivisible unit during a simulation, MD simulations can be roughly divided into two major categories: All-atom Molecular Dynamics and Coarse-grained Molecular Dynamics (CGMD):
-
All-atom Molecular Dynamics: each individual atom is treated as the smallest indivisible unit for motion and force calculations
-
Coarse-grained Molecular Dynamics: a set of adjacent atoms (such as an amino acid residue, a water molecule) is treated as a coarse grained unit, usually referred as a “bead”. Only interactions between beads are considered, while all intra-bead interactions are neglected during a CGMD. This treatment makes CGMD capable of performing simulations on a larger time scale and for larger physical systems with reduced cost of computation and increased loss of accuracy.
Depending on the accuracy of potential energy functions used during a simulation, MD simulations can be divided into three categories: Classical Molecular Dynamics (Classical MD, cMD), Ab-initio Molecular Dynamics (AIMD) and Machine Learning Molecular Dynamics (MLMD):
-
Classical Molecular Dynamics: potential energy functions of the physical system come from a force field;
-
Ab-initio Molecular Dynamics: potential energy functions of the physical system come from ab-initio calculations;
-
Machine Learning Molecular Dynamics: potential energy functions of the physical system come from a machine learning force field.
Potential Energy Function
Potential Energy Function, usually shortened as “Potential”, refers to the function that is used to describe the energy of interaction within a physical system. In an all-atom MD simulation the potential is a function of the atom types and atomic coordinates within the given physical system, and it could be given by quantum mechanics (QM), molecular mechanics (MM) force fields, or machine learning (ML) force fields.
Force Field
Force Field, conventionally called Molecular Mechanics (MM) Force Field, refers to a collection of empirical functions with fixed mathematical formats to describe the potential energy of the physical system. Parameters for these empirical functions are determined by fitting against experimental data or QM-derived data. Compared to ab-initio methods, MM force fields are less accurate but much faster (usually several magnitudes).
Hamiltonian
Under the context of classical mechanics, the concept of the Hamiltonian refers to the total energy of a physical system, which is the sum of the potential energy and the kinetic energy of all particles within the given system.
In quantum mechanics, the Hamiltonian should be considered as an Hamiltonian operator.
Statistical Mechanics
In physics, statistical mechanics is a sub-discipline which applies statistical methods and probability theory to describe large assemblies of microscopic particles so that macroscopic behavior of the physical system (for instance, temperature, pressure) can be related to the behavior of microscopic particles.
State Function
State Function is a physical property to describe the macroscopic property of a physical system. State functions have fixed values for a physical system under certain thermodynamic equilibria and depend only on the current equilibrium state of the system, rather than the path on which the system reaches equilibrium. Examples of State Functions include internal energy, enthalpy, entropy, free energy, etc.
Ensemble
Ensemble is a concept in statistical mechanics, which refers to a collection of a large number of independent systems with identical properties and structures in various motion states under certain macroscopic conditions.
Free Energy
The thermodynamic free energy refers to the energy of a thermodynamic system that can be used to do external work. It can be used as a criterion for whether a thermodynamic process can proceed spontaneously. Under given constraints, the system always tends to transition to a state with low free energy. For example, the process of protein folding is the spontaneous transition from an unfolded state with higher free energy to a folded state with lower free energy. According to the different qualifications, it can be divided into Helmholtz free energy (common notation \(F\)) and Gibbs free energy (common notation \(G\)). Note: free energy is different from potential energy although many people may confuse them.
Boltzmann Distribution
In statistical mechanics, the Boltzmann distribution describes the In statistical mechanics, the Boltzmann distribution describes the probability distribution of particles in a system in possible microscopic quantum states, and has the following form:
where \(E\) is the quantum state energy, \(k\) is the Boltzmann constant \(T\) is the temperature, \(p_i\) is the probability that the particle is in the \(i\) quantum state, and ε\(_i\) is the energy of the \(i\) quantum state.
Collective Variables (Reaction Coordinates)
The representative parameters that can quantitatively describe the change process of the system are called Collective Variables (CV) or Reaction Coordinates (RC). For example, in the chemical reaction shown in the figure below, the distance between O and C \(d(\mathrm{C-O})\) can be regarded as the reaction coordinate, and the distance between C and Br \(d(\mathrm{Br-C})\) can also be regarded as the reaction coordinate.
Given that the reaction coordinates are well defined, methods such as umbrella sampling can be used to estimate the free energy difference between different reaction coordinates through molecular simulation, and then the free energy change along with the reaction coordinates during the transforming process can be described, which is the basis of kinetic and thermodynamic research.
Slow Degrees of Freedom
In the process of dynamic simulation, some degrees of freedom change rapidly with time (such as bond length, bond angle, etc., usually on the order of fs or ps). And some degrees of freedom change slowly with time (such as the dihedral angle, usually on the order of ns, \(\mu\) s , or even ms).
Enhanced Sampling
Enhanced sampling refers to accelerating the sampling of slow degrees of freedom in the simulation process by some technical means, which are classified as collective variable-based (e.g. umbrella sampling), and collective variable-free (e.g. replica exchange).
Quantum Mechanics
Quantum Mechanics is a branch of physics that studies microscopic systems. By describing the motion and interaction of microscopic particles (such as electrons, protons, etc.), quantum mechanics can explain many experimental phenomena that cannot be explained under the framework of classical mechanics, including blackbody radiation and the spectrum of the hydrogen atom.
Operator
Generally, an operator acts on the state space of a physical system, making the physical system transform from one state to another. Within the context of quantum mechanics, the state of a system can be described by a state vector. Physical observables (such as position, momentum, Hamiltonian, etc.) all correspond to a (Hermitian) operator.
Schrödinger Equation
In quantum mechanics, the Schrödinger equation is a partial differential equation that describes the time evolution of the quantum state of a physical system and is the fundamental equation of quantum mechanics. The Schrödinger equation can be divided into two types: the “time-dependent Schrödinger equation”
and the “time-independent Schrödinger equation” (also known as the steady-state Schrödinger equation)
where \(\hat{H}\) is Hamiltonian operator, and Ψ is the wave function of the system.
The time-dependent Schrödinger equation describes how the wave function of a quantum system evolves over time, while the time-independent Schrödinger equation describes the physical properties of a stationary quantum system.
First Principle
First Principle, also called ab initio, refers to derivation and calculation based on the basic laws of physics without additional assumptions and empirical fitting. For example, the of use the Schrodinger equation to solve electronic structure.
Wave Function
In quantum mechanics, the state of a quantum system can be described by a wave function. The wave function \(Ψ(r,t)\) is a complex-valued function. According to Bonn’s statistical interpretation, \(\|Ψ\|^2\) is the probability density of finding a particle at position \(r\), time \(t\).
Born-Oppenheimer Approximation
The Born-Oppenheimer approximation refers to the approximate variable separation of the nuclear coordinates and the electron coordinates when solving quantum mechanical equations containing the nucleus and electrons, to decompose the wave function of the whole system into separately solving the nuclear wave function and the electron wave function, which are two relatively simple processes. The basis of this approximation is that the mass of the nucleus is 3 to 4 orders of magnitude larger than that of the electron, and the speed of the nucleus is much smaller than that of the electron, so the electron can be regarded as being in the potential field formed by the stationary nucleus, and the nucleus won’t be affected by the specific position of the electron, only the average force of electrons counts.
Density Functional Theory
Density functional theory (DFT) is a quantum mechanical method to study the electronic structure of multi-electron systems, and it is one of the most commonly used methods in the fields of condensed matter physics and computational chemistry. Since the classical method of electronic structure theory needs to solve the multi-electron wave function with a higher dimension (\(3N\) for a system containing \(N\) electrons), the basic idea of the density function is to use the electron density instead of the wave function as the basic amount of research, thereby reducing the computational complexity. The most common application of density functional theory is implemented with the Kohn-Sham method.
Chemistry
Atomic Orbitals
In Quantum Mechanics, Atomic Orbitals are mathematical functions that describe the wave-like behavior of electrons in atoms. This function can be used to calculate the probability of electrons appearing around the nucleus, and the meaning of “orbital” refers to the probability of electrons appearing in a specific area. According to the “shape” of the track, it can be classified into s, p, d, f, etc.
Electronegativity
Electronegativity describes the ability of atoms of an element to attract electrons in a compound. The greater the electronegativity of an element, the stronger the ability of its atoms to attract electrons in the compound. In a period of the periodic table, the electronegativity of the element atom increases from left to right; and it decreases from top to bottom in a group. Therefore, the elements at the upper right of the periodic table (O, N, F, Cl, etc.) have higher electronegativity values. The element with the greatest electronegativity is fluorine.
Chemical Bond
A chemical bond refers to the strong interaction between atoms, ions, and other particles. Through chemical bonds, particles can form polyatomic compounds (such as organic molecules, inorganic molecules, ionic compounds, etc.). Simply put, for a polyatomic system, the most stable configuration between positively charged nuclei and negatively charged electrons is that when electrons are located between nuclei, electrons are attracted between different nuclei, and using this force the nuclei are “attracted” together, forming a chemical bond.
-
Ionic Bond: A chemical bond formed by electrostatic interaction between oppositely charged anions and cations, without directionality, such as sodium chloride (salt), calcium carbonate.
-
Covalent Bond: A chemical bond formed by sharing electron pairs between atoms. Two atoms with similar electronegativity are equally attracted to electrons, so they mainly form chemical bonds by sharing each other’s outer valence electrons. Covalent bonds are directional, resulting in complex molecular structures. For example, in the methane molecule, carbon atoms and hydrogen atoms are connected by covalent bonds to form a regular tetrahedron, the carbon atom is located at the center of the tetrahedron, and the hydrogen atom is located at the vertex of the tetrahedron. According to the number of shared electron pairs, it can also be classified into a single bond, double bond, and triple bond.
-
Hydrogen Bond: When a hydrogen atom forms a covalent bond with an atom with high electronegativity X (usually O, N, F), if it bonds with another atom with high electronegativity. When Y (usually also O, N, F) is close, using hydrogen as the medium between X and Y, a special form of interaction like X-H· · ·Y is generated, known as a hydrogen bond. Hydrogen bonds widely exist in biological macromolecules such as water and proteins and DNA. It plays a crucial role in stabilizing the conformation of biological macromolecules.
Functional Group
Functional groups are atoms or groups of atoms that determine the properties of organic compounds. Common functional groups include hydroxyl (-OH), carboxyl (-COOH), ether bond (C-O-C), carbonyl (C=O), halogen atom (-F, -Cl, -Br, -I), etc.
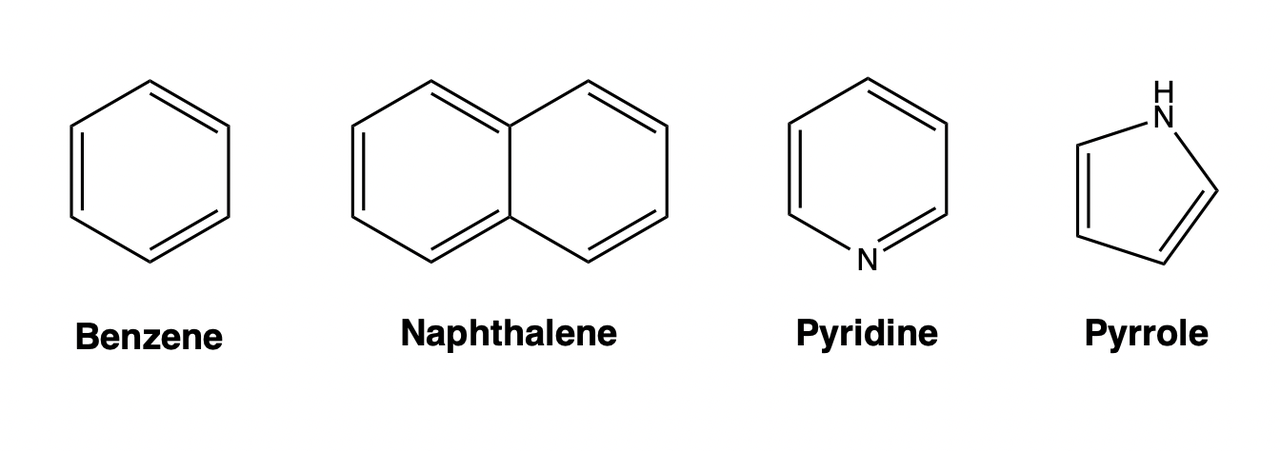
Aromatic
Aromaticity is a chemical property that exists in cyclic planar molecules co ntaining \(\pi\) bonds composed of delocalized electrons, which can provide molecules with stability that cannot be explained by conjugation alone. The number of electrons in the delocalized \(\pi\) of an aromatic molecule needs to satisfy the Huckel rule (also called the “4n+2” rule). Molecules with aromaticity are called aromatic compounds, and molecules without aromaticity are called aliphatic compounds. Aromatic compounds can be roughly classified into simple aromatic compounds (such as benzene), polycyclic aromatic compounds (such as naphthalene, and anthracene), and heterocyclic compounds (such as pyridine, and pyrrole).
Conformation
Conformer usually refers to three-dimensional conformation, which refers to the structure that a molecule has in three-dimensional space. For organic molecules, their conformations cannot be randomly generated due to the limitation of the directionality of covalent bonds.
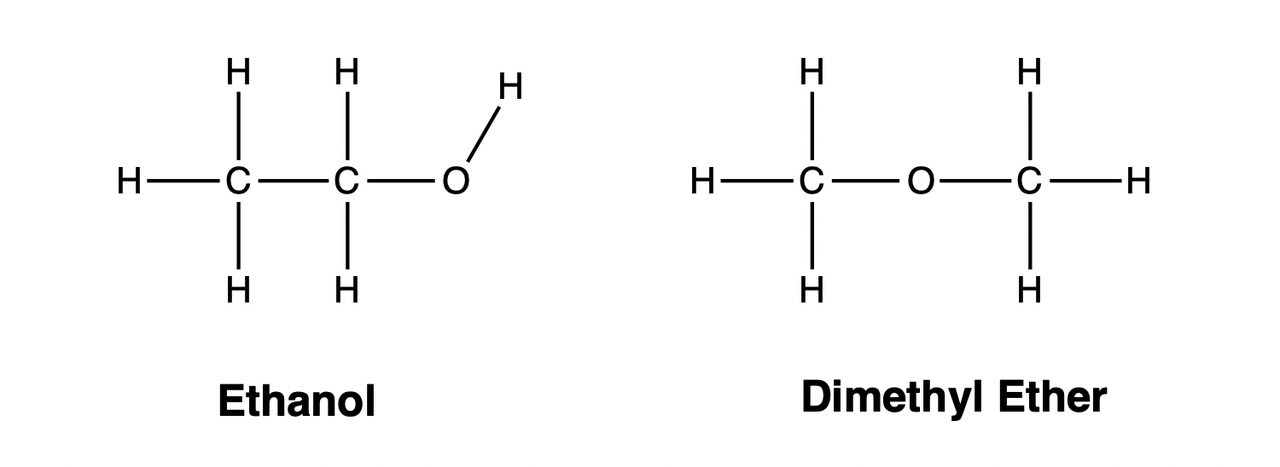
Isomers
In Organic Chemistry, substances with the same chemical composition (molecular formula) but different structures are called isomers of each other. For example, the compositions of ethanol and dimethyl ether are both \(\mathrm{C_2H_6O}\), but their structures are different:
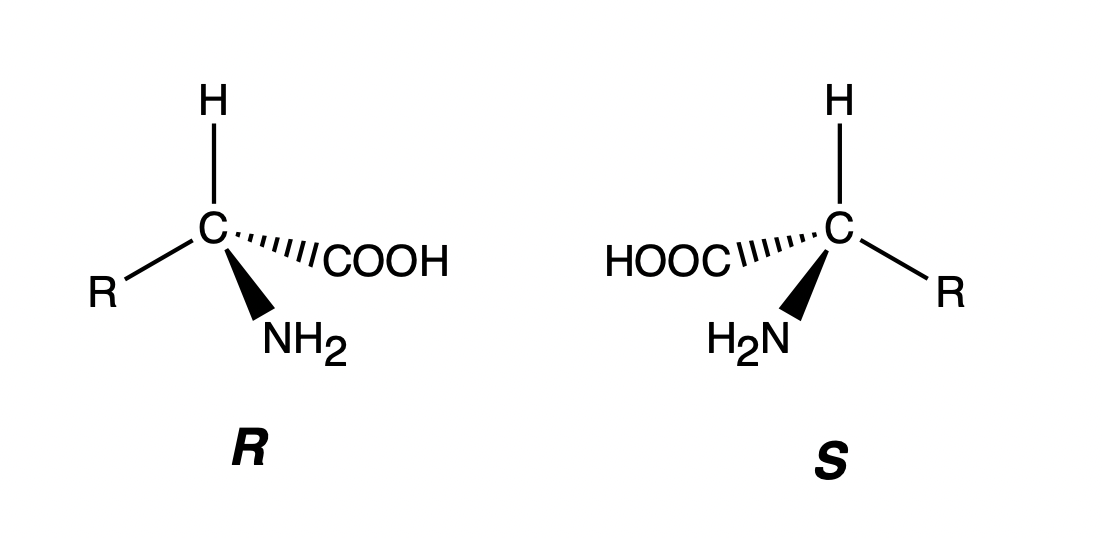
Stereoisomerism
Stereoisomers refer to molecules in which atoms are topologically connected in the same way but spatial arrangement of the atoms are different. For example, a molecule is likeley to have stereoisomers when it contains carbon atoms to which four different functional groups are bonded. Such atom is called chiral atoms, and usually R/S are denoted to distinguish two different them. In terms of biomolecules, such as peptides, amino acids and sugar, L/D are frequently used to denote different type of stereoisomers. The two amino acid configurations shown in the figure below are stereoisomers of each other. All natural amino acids are in the L configuration, and their carbon atoms are in the S configuration.
Cis-trans Isomerism
Cis-trans isomerism refers to isomerism that occurs due to the hindered free rotation in the compound molecule, which is commonly found in compounds with double bonds or rings.
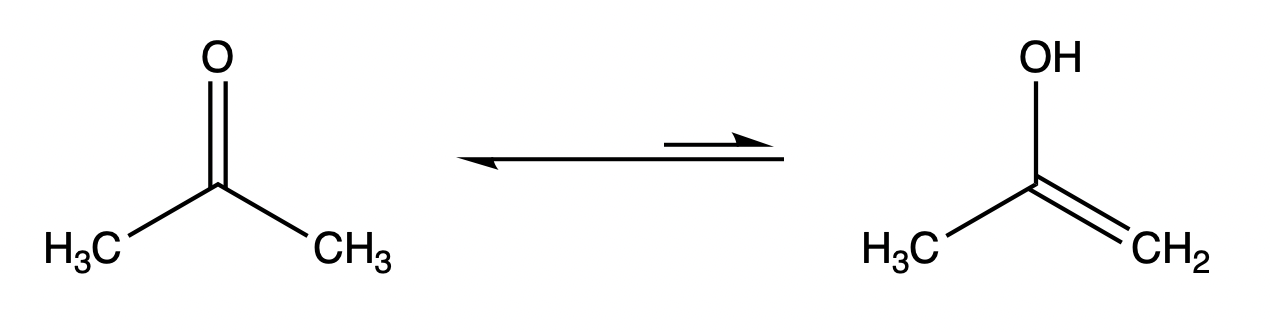
Tautomerism
Tautomerism means the structure of some organic compounds is converted between two functional isomers. Most tautomerisms involve the transfer of hydrogen atoms or protons, and the conversion of single bonds to double bonds. The distribution of tautomers in equilibrium depends on specific factors, including temperature, solvent, and pH, etc. The diagram below shows the keto (left) and enol (right) tautomers present in carbonyl compounds, with the keto structure predominating in the usual case.
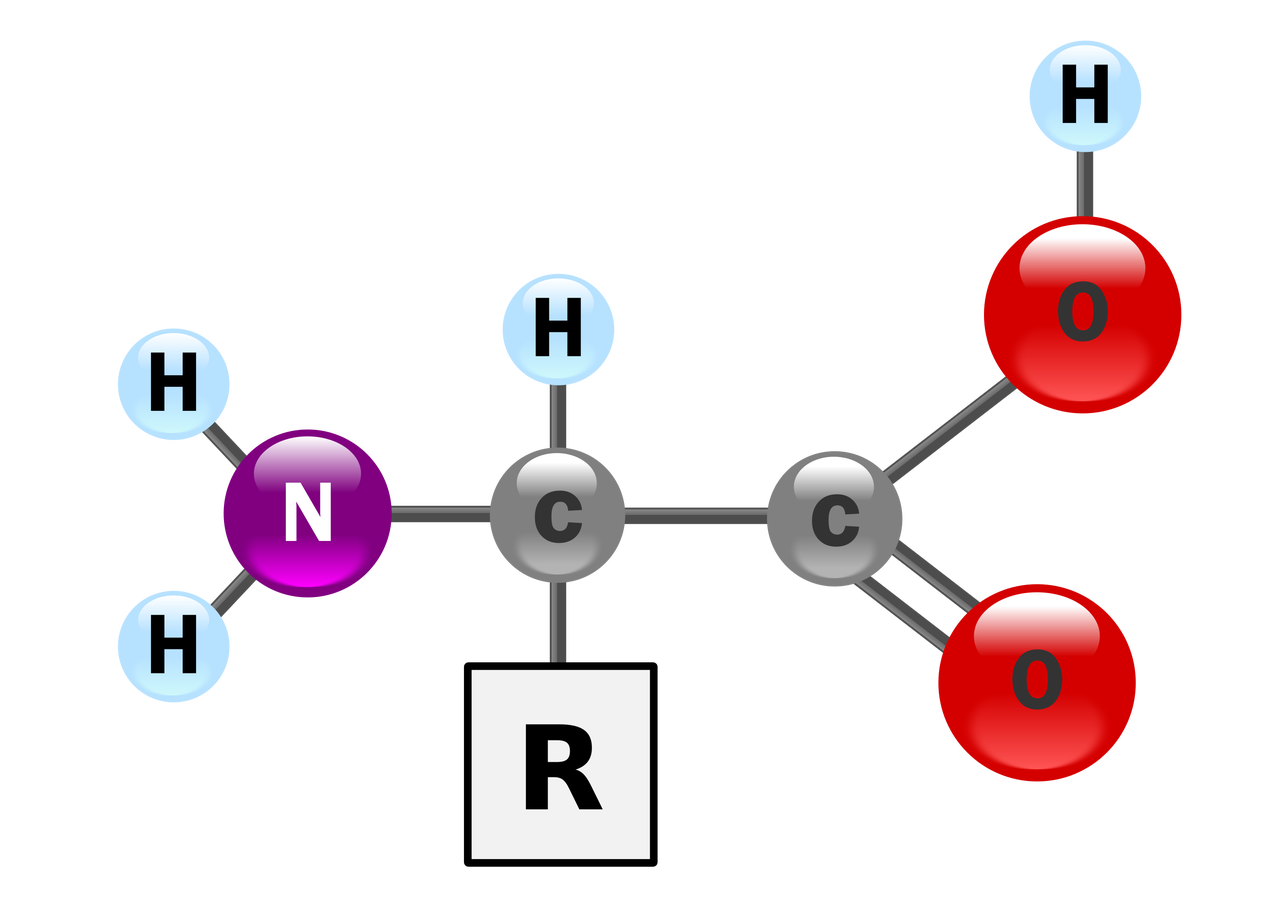
Amino Acids
Amino acids are biologically important organic compounds consisting of amino (-NH2) and carboxyl (-COOH) functional groups and side chains attached to each amino acid. Amino acids are the basic units that make up a protein. In nature, there are 20 genetically encoded amino acids.
Protein Structure
Protein structure refers to the spatial structure of a protein biomolecule, which can be divided into four levels to describe different aspects.
-
Primary structure: the linear amino acid sequence that makes up the polypeptide chain of a protein.
-
Secondary structure: a stable structure formed by hydrogen bonds between C=O and N-H groups between different amino acids, mainly \(\alpha\)-helix and β-sheet.
-
Tertiary structure: the three-dimensional structure of a protein molecule is formed by the arrangement of multiple secondary structural elements in three-dimensional space.
-
Quaternary structure: used to describe the interaction of different polypeptide chains (subunits) to form functional protein molecules.
Ligand
In biochemistry or pharmacology, a ligand refers to a compound that can bind to a receptor and then lead to some physiological effect. In medicinal chemistry, ligands are usually small organic molecules or short peptides composed of several amino acids. The forces between ligands and receptors are usually non-covalent interactions: such as hydrogen bonds, electrostatic interactions, van der Waals interactions, etc.
Receptor
Signal transduction is responsible for intracellular communication via series of molecular events (protein phosphorylation) upon chemical/physical signal outside cell , where receptor function in the central role as transmit signals outside cells and produce specific effects within cells. It is usually biological macromolecule such as protein. After the receptor binds to a specific stimuli, the structure will change to a certain extent, and the corresponding effect will be induced in the cell. In medicinal chemistry, receptors usually refer to target proteins able to bind with ligands.
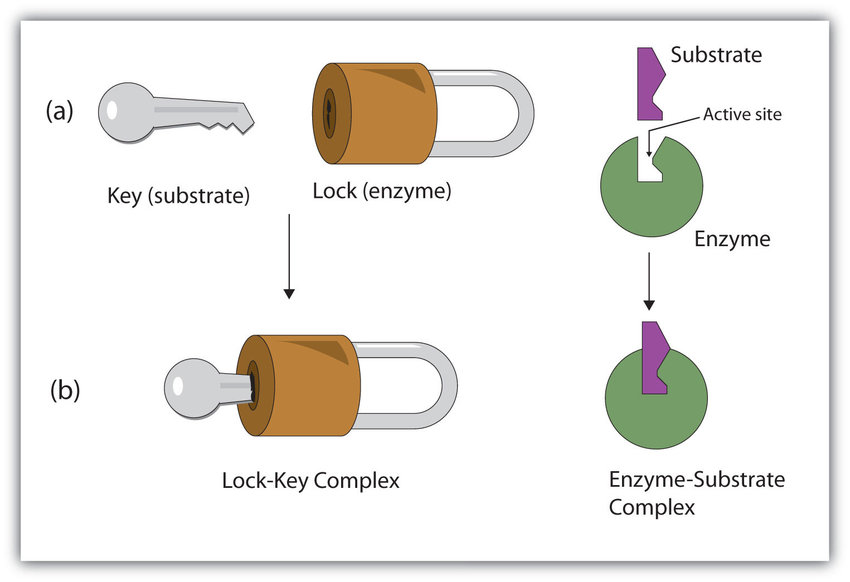
Lock and Key Model
The lock-and-key model is a theory proposed by E. Fischer in 1890 to explain the specific binding between enzymes and substrates (or between ligands and receptors). The model believes that the structures of enzymes and substrates at their binding sites should be strictly matched and highly complementary, just like the structural complementarity and matching of a lock and its original key. The disadvantage of this model is that the model treats the structure of the enzyme and the substrate as rigid structures, which is inconsistent with the fact that the conformation of the enzyme and the substrate changes during the catalytic reaction.
Induced Fit Model
The Induced-Fit Model is a model proposed by Koshland in 1958 to describe the enzyme-substrate (ligand-receptor) binding interaction. This model believes that in the process of binding the enzyme to the substrate, the substrate can induce a certain change in the structure of the enzyme, and finally form an active conformation that can bind to the substrate.
Molecular Docking
Molecular Docking is a technique that simulates the interaction between ligands and receptors. The technology predicts ligand binding modes and ligand-receptor binding forces by physically modeling intermolecular interactions and applying optimization algorithms such as the Monte Carlo method.
Reversible Reaction
A reversible reaction is a chemical reaction that can proceed in both the forward and reverse directions under the same conditions. When the degree of the reverse reaction direction is much smaller than that of the forward reaction direction, the reaction can be considered irreversible. Most of the reactions are reversible, such as the dissociation of weak acid/base, ligand-receptor binding, etc.
Chemical Equilibrium
Chemical Equilibrium refers to a state in which the forward and reverse reaction rates of a chemical reaction are equal in a reversible reaction with certain macroscopic conditions, and the concentrations of the reactants and the components of the products do not change. Take the following reaction as an example:
When the equilibrium is reached, the concentrations of \(\mathrm{A,B,C}\) are respectively [\(A\)],[\(B\)],[\(C\)], then the equilibrium constant K can be defined:
Given the reaction conditions, the equilibrium constant for a reaction with a fixed stoichiometric ratio is the same, and is related to the free energy change of the reaction as follows:
van der Waals force
van der Waals (vdW) force refers to the non-directional, unsaturated, weak interaction force between atoms. Van der Waals interactions are much weaker than chemical bonds, but they will significantly affect the melting point, boiling point, and many other properties. Van der Waals interactions have 3 major contributions:
-
Attractive or replusive interactions are between permanent charges, dipoles, quadrupoles, etc.
-
Induction (also known as polarization), which is the attractive interaction between a permanent multipole on one molecule with an induced multipole on another. This interaction is sometimes called Debye force.
-
Dispersion (usually named London dispersion interactions after Fritz London), which is the attractive interaction between any pair of molecules, including non-polar atoms, arising from the interactions of instantaneous multipoles.
In molecular simulations, van der Waals forces are usually described in terms of the Lanner-Jones potential function, which has the following form:
Where \(r\) is the distance between two atoms, \(C\) is a parameter, usually obtained by fitting physical quantities such as density and the enthalpy of evaporation.
Hydrophobic interaction
Hydrophobic interaction, also known as a hydrophobic effect, is a chemical phenomenon that which groups with hydrophobicity in an aqueous solution (such as alkyl groups without polarity) are close to each other to reduce the contact area with water. Hydrophobic interactions are the main driver of protein folding.
Thermodynamics
Thermodynamics focuses on the interaction of heat and work between chemical reactions and system states under the laws of thermodynamics. Generally speaking, the problems (equilibrium state) that do not involve the study of the chemical reaction process belong to the category of chemical thermodynamics, such as phase transition, and the balance of sodium and potassium ions on two sides of the cell membrane.
Kinetics
Kinetics, also known as reaction kinetics and chemical reaction kinetics, is a branch of physical chemistry that studies the rate and mechanism of chemical reactions. Chemical kinetics is different from chemical thermodynamics. It does not care about the equilibrium state, but studies the chemical reaction dynamically, and studies the time required for the transformation of the reaction system, as well as the microscopic process involved.
Biology
Receptor
Chemical groups (protein, in most of the cases, on the surface or inside cell) possessing the ability to bind with other chemical groups (with specificity). Receptors preferenced bounded by certain molecules (drugs) and involved in parthenogenesis of certain disease would be ideal targets for drug design.
Enzyme
An enzyme is a biological catalyst that is capable of accelerating a specific chemical reaction in cells. The enzyme is not destroyed during the reaction process and could be used again and again (under the sustained condition). In most cases, enzymes are proteins.
-
Active site Catalytic center of enzymes is the core region to bind substrate(s) and initiate reactions. For enzymes that are proteins, side chains along the backbone of key amino acids constructing the active site, shape it into specific size with specific chemical behavior. Given that an enzyme function in whole, sometimes it is also acceptable to simulate reaction mechanism focusing on the active site (while simplify the environment effects contributed from other parts of the enzyme as well as the solvent) by high-level quantum mechanics studies.
-
Cofactor/Prosthetic group/Coenzyme Cofactors are necessary non-peptide components required for enzymes to function properly. Cofactors can either be inorganic metal ions or organic molecules. The assistance of cofactors for enzyme function is achieved by binding to the inactive form of enzyme (apo-enzyme) to produce the catalytic active form (holo-enzyme). A prosthetic group is a type of cofactor that tightly bind to the assisted enzyme and is not easily to be removed. A coenzyme is a specific type of cofactor as they are organic small molecules.
-
Michaelis-Menten equationMichaelis–Menten kinetics describe the typical kinetic behaviour of enzymes. The name was given after German biochemist Leonor Michaelis and Canadian physician Maud Menten. The Michaelis–Menten kinetics model describes the rate of enzymatic reactions \(v\) in the form of Michaelis–Menten equation showing below:
Here, enzyme reaction rate \(v\), the rate of forming product \([P]\), is related with substrate concentration \([S]\). \(V_{max}\) describes the maximum reaction rate achieved by the studied system. It would be reached when the substrate concentration is saturated under a given enzyme concentration. The Michaelis constant \(K_{M}\) is numerically equal to the substrate concentration where half \(V_{max}\) is reached. In most of the enzyme catalyzing single-substrate reactions, their kinetics behaviours are assumed to fit Michaelis-Menten equation, regardless of further assumptions.
Phosphorylation
The process to add phosphoryl (\(PO_4\)) group(s) to molecules. In biological process, phosphorylation happens major in proteins and is catalyzed by kinases to transfer phosphoryl group from ATP (most of the times) to the target sites (amino acids, to be specific). The opposite process of phosphorylation is dephosphorylation, function by phosphatases to eliminate the phosphoryl group from residues. Phosphorylation status of proteins plays vital role in cell signalling pathway. Detection/prediction for protein phosphorylation (and phosphorylation site) is also a hot issue for state-of-art AI application.
Kinase
Types of enzyme responsible for substrate phosphorylation. Since they have been widely involved in tumorigenesis, kinase-targeting cancer therapies have attracted much attention. Among the kinase spectrum for cancer drug discovery, cyclin Dependent Kinases (CDK), anaplastic lymphoma kinases (ALKs), epidermal growth factor receptors (EGFR, types of receptor tyrosine kinases), janus kinases (JAKs) are included as the most popular studied ones.
- Receptor tyrosine kinase Tyrosine kinase is a type of kinase for tyrosine phosphorylation. It functions as an "on" or "off" switch in many cellular signalling process. Receptor tyrosine kinase is a subclass of tyrosine kinase that serves as cell surface receptor with high-affinity for many polypeptide growth factors, cytokines, and hormones.
Catalytic receptor
Type of cell surface protein with the ligand binding site localized at the extracellular surface of the plasma membrane and the functional region possessing catalytic activity on the intracellular face of the plasma membrane. The two parts are linked by a single transmembrane-spanning domain consisting of 20–25 hydrophobic amino acids. It commonly exists and functions as a dimer. Endogenous ligands for catalytic receptor are often peptides or proteins.
Transport protein
A transmembrane protein which function to allow selective passage of specific molecules from the external environment and is able to translocate ions, small molecules, or macromolecules. Transport proteins may be divided into subgroups as channels and carriers.
-
Ion channel An ion channel is a type of transmembrane protein that mediates the passage of ions through the membrane. The major differences between ion channels are ion carriers are: (1).high efficiency, usually \(10^6\) per second (or higher); (2).translocation of ions down their electrochemical gradient in an energy conservation way.
-
Carrier proteins Active carrier proteins function in the energy-consumed manner and are able to translocate the substances against concentration gradient. Passive carrier proteins assist the substance by facilitated diffusion.
Nuclear hormone receptors (NHR)
A class of transcriptional factors to regulate gene expression regulated by their binding ligands. The ligand binding domain (LBD) is capable of recognizing specific ligands to stimulate conformational change (dimerization) of NHR. The DNA binding domain (DBD) mediates the receptor towards its hormone response elements (HRE). DBD functions in the form of a dimer with each monomer recognizing a six base pair sequence of the targeted DNA. The peroxisome proliferator-activated receptor (PPAR) is a family of nuclear receptors involved in pathology of diabetes as well as cancer.
G protein coupled receptors (GPCR)
A large group of evolutionarily-related proteins serve as cell surface receptors to produce cellular response activation upon signal outside cell. The transmembrane domain of GPCRs pass through the cell membrane seven times (typical structure characteristics of GPCRs). Ligands can either bind at extracellular N-terminus and loops or within the transmembrane helices of GPCR. Effective binding of ligand would cause conformational change. Subsequent dissociation of \(\alpha\) subunit from the conjugated G-protein would further facilitate intracellular signal processing. GPCRs are widely targeted in drug discovery, as approximately 40% of marketed drugs modulate GPCRs.
Receptor conformation
It should be noted that proteins have flexibility and their conformation should be changed before and after ligand binding.
-
Apo form Conformation of a protein without any ligand bounded inside.
-
holo form Conformation of a protein with ligand bounded.
Binding site/Pocket
-
Allosteric site For enzymes (receptors) which possess allosteric regulation, in addition to casual ligand binding sites, called orthosteric sites, they also have the allosteric sites which could recruit effectors to bind and thus initiate conformational change to facilitate either enhanced activity or suppressed activity of the enzymes. Design allosteric modulators for medical use is one of the hotspot especially in aspects of treating CNS (Central nervous system) diseases. If one would like to know more about the allosteric regulators in real-world drug discovery (and try to create more crazy ideas with our beloved AI models), it would be a good start to search with target names as GPCRs, BCR-ABL, SHP2, JNK and CDK, to name a few.
-
Orthosteric site In scenario of allosteric regulation, the casual binding site of receptor is called orthosteric site. In some cases, it’s almost the same thing if one use ‘orthosteric site’, ‘ligand binding site’ as ‘active site’.
-
Cryptic site The "hidden" ligand binding site from receptors that should only be available and induced upon ligand binding. Typical receptor with cryptic site is Bcl-xL, which largely participants in cancer progression.
Apoptosis
Programmed cell death. The triggering factors of apoptosis could be cellular stress, DNA damage, immune surveillance, et.al. Since tumorigenesis is resulted from imbalance of proliferation and programmed cell death, therapy to target apoptosis and induce death of cancer cells has been widely carried out. The well known drug target in such way is the B-cell lymphoma 2 (BCL-2) family, which is in charge of controlling permeabilization of the mitochondrial outer membrane for apoptosis regulation.
Autophagy
Intrinsic recycling process of cellular components facilitated by lysosome-dependent degradation process. Although it is widely accepted for intimate relationship of autophagy with aging, detailed mechanism remains unclear. It is still a major process to target, when it comes to age-related pathologies, such as neurodegenerative diseases
Mitochondrial stress
Mitochondria is the energetic engine and signaling hub for eukaryotic cells. Mitochondrial stress is severe dysfunctional status triggered by metabolic stimuli and other changes, leading to mitochondrial unfolded protein response and a retrograde stress signaling pathway, resulting in endoplasmic reticulum stress and metabolic instability. Mitochondrial stress is highly involved in pathologies of cancer, age-related diseases as well as diabetes.
Ferroptosis
Recently discovered novel form for regulated cell death. Unique characteristics of ferroptosis is the initiating process of iron ion accumulation. Subsequent cell membrane damage is mediated by lipid peroxidation. As iron-dependent manners play vital roles in physiological properties of cancer cells, targeting ferroptosis indicates a promising direction for cancer drug discovery.
Ubiquitination
A biological process of protein degradation (intracellularly). The protein is first labelled with a ubiquitin, a 76-amino-acid protein, through a three-step process with help of ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin-protein ligase (E3), facilitating mono-ubiquitination. The labelled ubiquitin chain could be extended by adding more ubiquitin, resulting in polyubiquitination. The 26S proteasome (proteasome is the huge protein complex to break peptide bonds for unneeded or damaged proteins.) recognizes the polyubiquitination as a signal to initiate proteolysis and process the protein for degradation.
Heat shock proteins (HSP)
Molecular chaperones (proteins) to assist protein functioning in response to stressful conditions (eg. exposure to cold and/or UV light, wound healing etc). HSPs are named according to their molecular weight. HSP90 refers to HSPs which are 90 kilodaltons in size. Ubiquitin (8 kilodaltons) also possess heat shock protein features.
Cytoskeleton
- Tubline Structural unit for living cell skeletal system. Tubulins are proteins that can be polymerized into long chains or filaments to assemble into microtubules - hollow fibers that serve as cell skeletal system.
Cancer/Tumour immunology
-
Immunology and Immunotherapy Immunology studies the immune system of human body,A branch of physiology raising huge interest recently. Immunotherapy functions by activating/mediating/enhancing immune responses of the patients to fight against diseases (cancer).
-
Antigenicity and immunogenicity When a foreign material (antigen) enters, an organism would initiate a barrier system to fight against and eventually eliminate this intruder. Antigenicity describes the ability of an antigen bind to, or interact with the products of the final cell-mediated response (such as B-cell or T-cell receptors). Immunogenicity measures the ability of the antigen to activate the immune response (including innate immune response and the subsequent adaptive (acquired) immune response). Immunogens possess antigenicty, while antigens may not always have immunogenicity. Metal ions are typically haptens, which are antigens, but would not trigger immune responses.
-
Cluster of differentiation (CD) Surface proteins on leukocytes, reflecting differentiation stage or activation state of the cell and can be recognized by specific monoclonal antibodies. It is commonly to notice that CD4 lymphocyte labelled as CD4+ and CD8 lymphocyte as CD8+, the former is known as the T-helper cell and latter includes killer T cell.
-
Cytokine and cytokine storm Cytokines are messenger proteins released from immune cells to regulate immune responses. Abnormal activities of cytokines could induce "cytokine storm" which has lethal impact.
-
Monoclonal antibody Monoclonal antibodies are engineered antibodies that typically recognize the same epitope, and thus possesses high specificity towards the targeted antigen.
-
Vaccine Vaccines are the biological preparation containing an agent to initiate the immune responses to form a barrier thus protect the body from certain disease derived from infection. The agent of vaccine resembles the disease-causing microorganism and is often made from weakened or killed forms of the microbe, its toxins, or the surface proteins.
-
Humanized antibody Processing the antibody which is generated from non-human species with for example, recombinant DNA to create "insertion" into the antibody molecule. The original prepared antibodies possess partially different protein sequences from which are naturally occurred in human thus potentially suffer from being recognized as immunogenic once administered.
-
Single cell sequencing Perform sequencing for individual cells. It provides fundamental accesses to understand cell functionality and differentiation with higher resolution. Benefit from such novel technique on the basis of next-generation sequencing, early diagnosis of cancer from small populations of cells carrying certain mutations driven cancer prevention towards big progression.
-
CAR-T and TCR-T Chimeric Antigen Receptor (CAR-T) (cell) therapy functions by engineering T-cells of patients to fight against cancer. T-cells of particular patients are collected and engineered to express the chimeric antigen receptor protein on cell surface, which is responsible for recognition of cancer cells. These customized T-cells would be amplified and transferred back to patient’s body to recognize and kill cancer cells, thus serving as "living drugs". Different from CAR-T, TCR-T cell therapy focuses on the T cell receptor (TCR) located on surface of T-cells with genetic engineering program to enable wide recognition spectrum towards peptides that could be presented by human leukocyte antigens (HLA).
-
PD-1/PD-L1 As the well known checkpoint, programmed death protein 1 (PD-1) and its ligand PD-L1 (pair) mediate pathway largely involved in immunotherapy. PD-1 participants in the immunosuppressive pathway and is responsible for down-regulation of immune reaction. Binding of PD-L1 could inhibit the proliferation of PD1-positive cells. Overexpression of PD-L1 on surface of tumor cells cause cancer immune evasion and failure of treatment. Thus, blocking the contact of PD-1 and PD-L1 could suppress the immune suppression, enhance T cell responses and boost anti-tumor activity of treatments.
-
Neoantigen Neoantigens are the translation product (protein) of mutated DNA in cancer cells. They are different from the original protein under physiological condition and may thus play a significant role in stimulating immune response against cancer cells.
AI advances - Fundamental basis for rational drug design
-
Protein structure determination One of the most significant advancements in science in the 21st century is the application of AI in protein structure determination. The conventional method of determining protein structures through experimental studies is time-consuming and may not provide sufficient information for in-depth analysis. Homology modeling, which is based on the similarity between a protein and its related proteins with known atomic-level information, can provide some insight into protein structures but may not capture specific differences between the proteins. This presents a challenge for the pharmaceutical industry in terms of rational drug design. AI algorithms, such as AlphaFold, offer a solution to this problem by providing a means to approach the mysterious algorithms that encode the tertiary structure of proteins, which is crucial for their proper function. AlphaFold has demonstrated enhanced simulation speed and outstanding performance in structure determination, which aligns well with experimentally determined structures. This has broadened the target scope for drug discovery, as it can provide reasonable monomer structures and complexes, including homo/hetero-multimers and protein-peptide complexes.
-
Binding site determination The determination of the binding site(s) on a protein is crucial for drug discovery and development. The proper 3D structure of a protein, particularly potential drug targets, must be analyzed to determine if there are binding pockets for ligands to bind to with preferred affinity. Experimental methods, such as yeast two-hybrid system, mutational analysis, protein microarrays, X-ray crystallography, and mass spectrometry, are commonly used to detect binding sites. However, the utilization of AI algorithms could potentially provide a more efficient and accurate evaluation.
-
Protein-ligand interaction Following the determination of the binding site, the analysis of protein-ligand interaction is the next logical step. This analysis determines the fit of the proposed ligand, such as a small molecule, into the binding site and the strength of the interaction. The interaction format, polar or non-polar, and the driving forces behind the interaction, such as hydrogen bonding, must also be understood to determine the effectiveness and non-toxicity of the ligand as a drug candidate. Experimental methods, such as cryoEM, site-mutation, and docking and free energy calculation, can provide answers to these questions, but are often time-intensive and may suffer from limitations in accuracy. AI algorithms that can accurately and quickly evaluate the interaction pattern from known complexes can accelerate the evaluation process. There are huge lots of entry points to start your AI4S work in this area, try to start with proposing ligand binding poses fast and accurate.
-
When dynamic factor matters It is important to remember that proteins and receptors are not static and algorithms built on static models may have limitations. For example, binding site prediction models based on surface descriptors may not identify cryptic sites. To address this issue, algorithms that consider dynamic factors, such as RiD-kit, the reinforced dynamics toolkit, have been developed to enhance performance and capture rare events.
-
Protein-protein interaction (PPI) Protein-protein interaction (PPI) is crucial for proper biological processes. Specific recognition between interacting proteins is established through physical contact, driven by forces such as electrostatic interaction, hydrogen bonding, and hydrophobic effects. Based on these forces, recognition patterns in protein sequences, such as conserved regions of similar physicochemical properties, have been observed in certain types of PPI. By summarizing these interaction patterns, they can be used to predict biological effects and design functional protein sequences with desired bioactivity.
Pharmacy
Drug targets
Molecules that are intrinsically associated with particular diseases and could be specifically addressed by drugs to take action. Most of the known drug targets are proteins.
Medical chemistry
Medicinal chemistry refers to designing and synthesizing small molecules as a pharmaceutical agent.
-
Hit-lead-candidate: A hierarchical description to describe the potential precursor of a registered medication entity.
-
Hit: Promising candidates from preliminary screening, typically with a micro-molar EC50 (median effect concentration) values, or top scores from virtual screening.
-
Lead: Candidates demonstrating further potential to become drugs. Lead compounds normally require extensive modification and assessment before becoming a drug candidate.
-
Drug candidate: A well-studied compound, showing sufficient evidence in potency, selectivity, safety, and other drug-like properties. Drug candidates will become registered drugs after thorough clinical trials(usually including thousands of volunteers, tens of years, and investment in the order of billions of dollars)
Druggability
If a protein is suitable to be a drug target. Studies on druggability don’t focus on ‘determination of a good target’, but rather on ‘how to filter out the unsuitable/difficult targets’. Two ‘tangible’ sub-project in this topic: if a target can be modulated by small molecules (containing suitable pocket / covalent modification site / etc for molecule binding); if the inhibition / activation of a target can cause downstream (at least cellular-level) changes (rather than be antagonised and eliminated due to intracellular homeostasis)
Bioactivity
Bioactivity refers to the fraction (%) of an administered drug that reaches systemic circulation.
Pharmacokinetics
Pharmacokinetics studies how organisms process drugs.
-
Absorption (A) studies how drugs get into the bloodstream.
-
Distribution (D) studies how drugs are reversibly transferred from one location to another within the body. some drugs tend to concentrate in part of the body like adipose tissue, raising potential risks for clinical usage.
-
Metabolism (M) studies how drugs are broken down and modified inside the body.
-
Excretion (E) studies how drugs and their metabolites (a metabolised form of drugs) are removed from the body.
-
Toxicity (T) is a pharmacodynamic property of drugs. Since its assessment protocol shares something similar with ADME, they are referred to as a whole in many cases.
Pharmacodynamics
Pharmacodynamics studies what a drug does to the body. Pharmacodynamics focus on the molecular, biochemical and physiological effects or actions of the studied drug.
Phase I/II biotransformation
metabolism of a drug can be divided into 2 phases. Phase I mainly involves the breakdown (mainly by hydrolysis and oxidation). Phase II mainly involves the conjugation of chemical groups (polar in most cases) to make drug more soluble and suitable for excretion.
-
Cytochrome P450 A family of key enzymes contain heme as the cofactor to function as mono-oxygenases. It is the typical phase I drug metabolizing enzyme and are involved in so many components’ metabolism from drug and food. They can be easily induced and inhibited by their substrate thus have a outstanding role when studying the drug-drug interaction (DDI). e.g. Patients who are taking Alvastatin are not allowed to eat grapefruit.
-
UDP-glucuronosyltransferases (UGT) A family of enzymes capable of transferring glucuronic acid group from uridine diphosphate glucuronic acid (the donor) to substrates thus form the conjugated products facilitating excretion. UGT is the number one enzyme family for phase II biotransformation.
-
Drug-drug interaction(DDI) It is commonly to see the co-prescription of medicines to enhance, co-operate in treating certain disease since it is often a syndrome. Thus it is also commonly to aware if co-admission would cause conflict for safety concern. Typical scenario would be that one effective medical entity blocks the metabolism pathway of another, say becomes an inhibitor of certain isoform of phase I/II biotransformation enzymes, which happen to be the one that is responsible for metabolic clearance of another co-prescribed medical entity, causes unusual accumulation of the entity and triggers the risk affairs. In this way, to get rid of the potential risks of co-prescription (and endogenous impact caused by drug administration), metabolism properties of the studied medical entity should be comprehensively understood during the development process (pre-clinical phase).
AI acceleration advances
Druggability Prediction for Receptors
The druggability prediction for receptors aims to determine whether certain receptors can serve as drug targets that can be specifically targeted by a drug for the treatment of a specific disease. Additionally, it is common to assess whether newly discovered receptors can be targeted by existing drugs for repurposed therapeutic applications (under the circumstances of drug repurposing/repositioning)
-
Pharmacophore (according to IUPAC) refers to an ensemble of steric and electronic features that are essential to ensure optimal supramolecular interactions with a specific biological target and to trigger (or inhibit) its biological response. In essence, during a quantitative study, a portion of a molecule is considered as a group, and it is described by its chemical properties (charge, hydrophobicity, aromaticity, steric hindrance, etc.). This group is then scored and replaced as a whole.
-
Structure-activity relationship (SAR) refers to the relationship between the structure and the biological activity of a compound. It attempts to address two questions: 1) which parts of a bioactive compound, or which combination of these parts, are important (specific pharmacophores in a certain topological/geometrical structure); 2) how to modify a molecule based on the information obtained above (inferring a stronger pharmacophore/scaffold to replace the previous one).
-
Drug similarity Similarity is a crucial metric in drug design, especially in the context of ligand-based drug design (SBDD). Similarity can be detected, defined, or compared based on chemical elements and/or bond order involved in entity formation, spatial shape, and properties derived from its 3D structure (charge density map, etc.), as well as the interaction mechanism of the molecule resembling that of a reference molecule (which could be a common drug molecule or a promising hit molecule). It is hypothesized that the more similar a search molecule is to the reference molecule, the more likely it is to serve as a drug (or drug candidate) like the reference.
-
Bioisostere refers to chemical substitutions or groups with similar physical or chemical properties that produce broadly similar biological effects. The replacement of bioisosteres from one compound to another is mainly employed when the parent compound is unsuitable for safe use, and/or bioavailability, etc., while still possessing desirable druggability characteristics.
Patent recognition/Literature information extraction
Patent recognition and literature information extraction are important topics in the field of natural language processing. A recent research in this area showed that natural language processing methods can be effective for information extraction from chemical patents (ChEMU 2020). Despite its significance, this area is often overlooked in the molecular machine learning literature.
Virtual screening
Virtual screening is a computational method for evaluating the potential of small molecule compounds as drug candidates by comparing their structures and properties to a target protein. This approach can help to reduce the time and cost of the drug discovery process.
Molecule Generation
Design a brand new chemical entity satisfying all demands (have ideal property, can be synthesised easily, haven’t been patented) is considered as the holy grail of drug discovery. Since the inference of the above properties is still underdeveloped, there is still a long way to go for this ambition. However, today’s development of generative chemistry models can also serve a practical role in settings like library generation (generate at least novel and ‘drug-like’ molecule) and conditional design (generate molecule satisfying certain explicit constraint).
-
De novo generation One of the main challenges of de novo generation is balancing the exploration of the global solution with the exploitation of local minima in order to find ideal topologies with druggability. De novo generation can start from atoms, fragments, or building blocks that are ready for chemical reactions. AI-based generative models commonly use deep learning architectures, such as variational autoencoders (VAEs) or generative adversarial networks (GANs), to learn the patterns and syntax embedded in diverse training structures and a generator that meets specific generation goals through bayesian optimization and reinforcement learning. It is important to remember in the pharmaceutical industry that the most important factor is not the performance of the model or the theoretical significance of the algorithm, but rather the generated structures must be easily synthesizable and effective. The limited perspective of de novo generation, as derived from the literature, includes strict consideration of synthesizability parameters, efficient sampling from high-standard building block libraries for constrained generation within preferred local minima wells, consideration of the interaction environment, such as the target binding site and preferred interaction format, and the addition of gene expression signatures to facilitate efficient hit discovery based on transcriptomic profiles.
-
Fragment based drug design (FBDD) is another approach to drug discovery that is becoming increasingly popular. FBDD uses small molecular fragments as starting points for drug design and has been shown to be effective in various studies. In comparison with conventional virtual screening which processing the whole molecules for search, FBDD screens the building blocks for specific targets and accumulates candidate fragments acting as starting points to be developed into leading compounds. In addition to reduce dimensions when travelling through chemical space, FBDD could also help to combine attractive properties that the screen-out fragments have, such as preferential binding affinity towards single isoform among large protein family, favorable physical and pharmacokinetics/toxicity properties. Upon building of reasonable fragment libraries, screening process embeded in FBDD mainly rely on biophysical techniques, take differential scanning fluorimetry, surface plasmon resonance, and thermophoresis as examples. Large majority in silico approaches boost the follow-up fragment-to-lead optimization by: growing, linking and merging strategies. It is widely required for AI involved techniques to (1).facilitate high-standard fragments library from which lead compounds could be recognized with higher possibilities; (2). integrate hit fragments largely preserve the positive synergistic effects.
Molecule synthesis
-
Reaction Output Prediction The prediction of the product(s) from the given reactants requires a comprehensive understanding of reaction rules and preference reaction sites. Organic reactions are inherently complex and can result in different outcomes due to factors such as the reaction conditions and the presence of multiple reaction sites in a single reactant. It is crucial to study and consider these variables to accurately predict the reaction output.
-
Retrosynthesis Retrosynthesis is a strategy used to design a synthesis pathway for target organic molecules. This method involves breaking down the target into simpler precursors and continuing this process until the building blocks are commercially available. This approach allows for a systematic design of the synthesis pathway.
-
Synthetic Route Design Synthetic route design involves planning and designing the most efficient synthetic route for a promising drug candidate. The aim is to find a suitable starting material and process that will ultimately yield the desired product. This step is crucial in the drug discovery process and can significantly impact the efficiency and cost-effectiveness of the overall synthesis process.
A fairy tales
If you ever wondering is there any drug discovered/designed by AI, the answer is yes. Top tier clinical stage AI-driven drug R&D company, Insilico Medicine announced the first preclinical drug candidate INS018-055 that is designed by AI targeting AI discovered target for idiopathic pulmonary fibrosis (IPF). The phase I trail of INS018-055 in New Zealand has reached positive topline results. The whole R&D process of INS018-055 takes only 18 months and the embedded end-to-end AI platform has been proved to be solid enough for real world drug discovery. It is not something in our dream to use AI for drug discovery, it’s true and happening.